Research Area: Network Analysis & Data Integration
We develop bioinformatics methods and implement user-friendly software to integrate and analyze networks of interacting molecules with respect to data quality and topological criteria as well as functional, structural, and evolutionary aspects. In particular, we study the role of proteins and their complexes in dynamic molecular networks.
Our research work has recently focused on thorough evaluations of publicly available datasets of human protein interactions and expression data. Our in-house data warehouse integrates the biological information on human proteins and their interaction and functional associations contained in diverse experimental and computational datasets. Currently, we develop and apply a comprehensive analysis pipeline for viral RNA interference data together with members of the ViroQuant project in Heidelberg.
Part of the integrated biological information is now available in our novel searchable BioMyn resource for human systems biology and medicine. Recently, we used it successfully for finding human disease proteins as well as dozens of scaffold proteins in interactomes. Scaffold proteins play an important functional role in many signaling pathways, mediating the assembly of signal-processing protein complexes.
In addition, we developed the new online information system PSISCORE, which complements the PSICQUIC protocol to exchange molecular interaction data with confidence scores. Multiple scoring servers can be used to assess the quality of individual interactions. PSISCORE succeeds our prototypic DASMI system, a distributed scoring approach based on a decentralized client-server architecture.
In addition, our web service FunSimMat and the Cytoscape plugin NetworkAnalyzer have already attracted a large user community. FunSimMat provides various important measures for the functional similarity of genes or proteins annotated with Gene Ontology terms. NetworkAnalyzer allows the efficient computation and visualization of many topological network parameters like the degree distribution or shortest path lengths. Recently, a collaborating research group has applied it for studying networks of polyanion-binding proteins in yeast and human.
Contact: Dr. Mario Albrecht (E-mail: mario.albrecht@mpi-inf.mpg.de)
Protein-protein interactions (PPIs) are fundamental for almost all cellular processes. The identification of PPIs reveals functional relationships between proteins and uncovers proteins involved in human diseases. Currently, the complete human protein interactome is estimated to contain up to half a million interactions. Most of these interactions are the result of small-scale experiments, but high-throughput initiatives have recently provided several thousands of new human interactions using the yeast two-hybrid technique or mass spectrometry. However, since the experimental coverage of the human interactome is still low, various bioinformatics methods have been developed to predict PPIs.
We compared predicted datasets, high-throughput results, and manually curated interactions taken from the literature. We primarily assessed the quality and the potential bias of the different datasets using (i) a new functional similarity measure based on the Gene Ontology, (ii) structurally known protein domain interactions, (iii) likelihood ratios, and (iv) topological network parameters. Our analysis results provide important insights into the qualitative differences between experimental and predicted data. In particular, our findings enable the derivation of appropriate confidence scores for the selection of reliable interactions and functional hypotheses, which form the basis of further studies, for instance, on network perturbations underlying diseases.
Figure:
Recall-precision plots using (A) the manually curated human protein
reference database (HPRD) or (B) two combined yeast two-hybrid datasets as
positive reference sets. Circles denote predicted datasets of PPIs, diamonds
high-throughput data, squares manually curated interactions taken from the
literature, and triangles randomized interaction datasets.
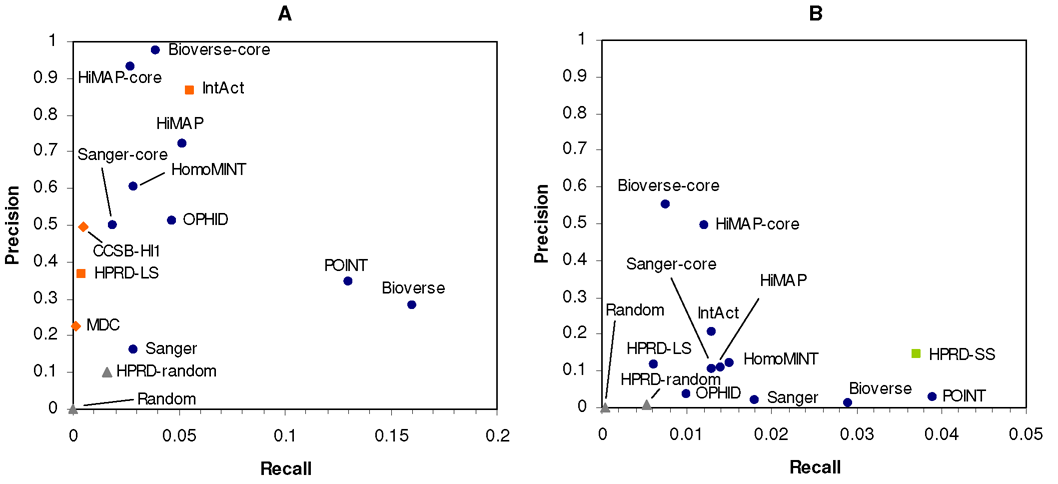
Our analysis revealed major differences between predicted datasets, but some of them also scored at least as high as the experimental ones regarding multiple quality measures. Therefore, since only small pairwise overlap between most datasets is observed, they may be combined to enlarge the available human interactome data. For this purpose, we additionally studied the influence of protein length on data quality and the number of disease proteins covered by each dataset. We could further demonstrate that protein interactions predicted by more than one method achieve an elevated reliability. We also find that experimentally derived interactions are prone to false positives, but, with the use of appropriate filters, subsets of elevated confidence can be derived.
Bioinformatics Cooperation Partners
- Dr. Henning Hermjakob, European Bioinformatics Institute, Hinxton, United Kingdom.
- Prof. Dr. Attila Reményi, Department of Biochemistry, Eötvös Loránd University, Budapest, Hungary.
- Prof. Dr. Petra Mutzel, Department of Computer Science, Dortmund University of Technology, Germany.
- Prof. Dr. Jörg Rahnenführer, Department of Statistics, Dortmund University of Technology, Germany.
- Members of the European BioSapiens Network of Excellence.
References
- Aranda, B., Blankenburg, H., Kerrien, S., Brinkman, F.S., Ceol, A., Chautard, E., Dana, J.M.,
De Las Rivas, J., Dumousseau, M., Galeota, E., Gaulton, A., Goll, J., Hancock, R.E., Isserlin, R.,
Jimenez, R.C., Kerssemakers, J., Khadake, J., Lynn, D.J., Michaut, M., O’Kelly, G., Ono, K.,
Orchard, S., Prieto, C., Razick, S., Rigina, O., Salwinski, L., Simonovic, M., Velankar, S.,
Winter, A., Wu, G., Bader, G.D., Cesareni, G., Donaldson, I.M., Eisenberg, D., Kleywegt, G.J.,
Overington, J., Ricard-Blum, S., Tyers, M., Albrecht, M., Hermjakob, H.
PSICQUIC and PSISCORE: accessing and scoring molecular interactions.
Nature Methods, 8(7):528-529, 2011.
(Abstract) (Supplement) - Emig, D., Albrecht, M.
Tissue-specific proteins and functional implications.
Journal of Proteome Research, 10(4):1893–1903, 2011.
(Abstract) (Supplement) - Emig, D., Kacprowski, T., Albrecht, M.
Measuring and analyzing tissue specificity of human genes and protein complexes.
EURASIP Journal on Bioinformatics and Systems Biology, 2011(1):5.1-6, 2011.
(Abstract) (First presented and published in Proceedings of the 7th International Workshop on Computational Systems Biology (WCSB), Luxembourg, 2010, in
Tampere International Center for Signal Processing (TICSP series), Tampere University of Technology, Tampere, Finland, ISBN 978-952-15-2384-7, 51:27-30, 2010.) - Ramírez, F., Albrecht, M.
Finding scaffold proteins in interactomes.
Trends in Cell Biology, 20(1):2-4, 2010.
(Abstract) (Supplement) - Blankenburg, H., Finn, R.D., Prlić, A., Jenkinson, A.M., Ramírez, F., Emig, D., Schelhorn, S.E., Büch, J., Lengauer, T., Albrecht, M.
DASMI: exchanging, annotating and assessing molecular interaction data.
Bioinformatics, 25(10):1321-1328, 2009.
(Abstract) (Supplement) (Press release) - Blankenburg, H., Ramírez, F., Büch, J., Albrecht, M.
DASMIweb: online integration, analysis and assessment of distributed protein interaction data.
Nucleic Acids Research, 37(Web Server issue), W122-W128, 2009.
(Abstract) (Press release) - Frishman, D., Albrecht, M., Blankenburg, H., Bork, P., Harrington, E.D.,
Hermjakob, H., Jensen, L.J., Juan, D.A., Lengauer, T., Pagel, P., Schachter, V., Valencia, A.
Protein-protein interactions: analysis and prediction.
In Modern Genome Annotation: The Biosapiens Network, Springer-Verlag, Wien, Austria, ISBN 978-3-211-75122-0, 353-410, 2009.
(Abstract) - Assenov, Y., Ramírez, F., Schelhorn, S.E., Lengauer, T., Albrecht, M.
Computing topological parameters of biological networks.
Bioinformatics, 24(2):282-284, 2008.
(Abstract) (Supplement) (First presented at the 5th Annual Cytoscape Retreat, Amsterdam, The Netherlands, 2007.) - Schlicker, A., Albrecht, M.
FunSimMat: a comprehensive functional similarity database.
Nucleic Acids Research, 36(Database issue):D434-D439, 2008.
(Abstract) - Jenkinson, A.M., Albrecht, M., Birney, E., Blankenburg, H., Down, T., Finn, R.D.,
Hermjakob, H., Hubbard, T.J., Jimenez, R.C., Jones, P., Kähäri, A., Kulesha, E.,
Macías, J.R., Reeves, G.A., Prlić, A.
Integrating biological data - the Distributed Annotation System.
BMC Bioinformatics, 9(Suppl. 8):S3.1-7.
(Abstract) (First presented and published at the 5th International Workshop on Data Integration in the Life Sciences (DILS), Evry, France, 2008.) - Ramírez, F., Schlicker, A., Assenov, Y., Lengauer, T., Albrecht, M.
Computational analysis of human protein interaction networks.
Proteomics, 7(15):2541-2552, 2007.
(Abstract) (Supplement) (In this issue) (Press release) - Schlicker, A., Huthmacher, C., Ramírez, F., Lengauer. T., Albrecht, M.
Functional evaluation of domain-domain interactions and human protein interaction networks.
Bioinformatics, 23(7):859-865, 2007.
(Abstract) (Supplement) (First presented and published in Proceedings of the German Conference on Bioinformatics (GCB), Tübingen, Germany, 2006.
GI-Edition - Lecture Notes in Informatics (LNI), Köllen Verlag, Bonn, Germany, ISBN 978-3-88579-177-5, P-83:115-126, 2006.) - Tress, M.L., Martelli, P.L., Frankish, A., Reeves, G.A., Wesselink, J.J., Yeats, C., Ólason, P.Í., Albrecht, M.,
Hegyi, H., Giorgetti, A., Raimondo, D., Lagarde, J., Laskowski, R.A., López, G., Sadowski, M.I., Watson, J.D.,
Fariselli, P., Rossi, I., Nagy, A., Kai, W., Størling, Z., Orsini, M., Assenov, Y., Blankenburg, H.,
Huthmacher, C., Ramírez, F., Schlicker, A., Denoeud, F., Jones, P., Kerrien, S., Orchard, S.,
Antonarakis, S.E., Reymond, A., Birney, E., Brunak, S., Casadio, R., Guigó, R., Harrow, J.,
Hermjakob, H., Jones, D.T., Lengauer, T., Orengo, C.A., Patthy, L., Thornton, J.M., Tramontano, A., Valencia, A.
The implications of alternative splicing in the ENCODE protein complement.
PNAS, 104(13):5495-5500, 2007.
(Abstract) (Supplement) - Salamat-Miller, N., Fang, J., Seidel, C.W., Assenov, Y., Albrecht, M., Middaugh, C.R.
A network-based analysis of polyanion-binding proteins utilizing human protein arrays.
The Journal of Biological Chemistry, 282(14):10153-10163, 2007.
(Abstract) (Supplement) - Salamat-Miller, N., Fang, J., Seidel, C.W., Smalter, A.M., Assenov, Y., Albrecht, M., Middaugh, C.R.
A network-based analysis of polyanion-binding proteins utilizing yeast protein arrays.
Molecular & Cellular Proteomics, 5(12):2263-2278, 2006.
(Abstract) (Supplement)